Essay
Computer simulation in biochemistry. With the development of
both theory and computer technology, simulation has become a viable
way of investigating the behavior of biomolecules. First applied to
proteins 25 years ago by McCammon and coworkers, the method of Newtonian
molecular dynamics has now entered the mainstream of biochemistry. Recently,
mesoscale and multiscale methods have emerged, with the promise of bringing
up the scale of simulations from molecular level to organellar and even
cellular level.
In my dissertation work, I applied both types of models to answer questions
in neurobiology. Molecular dynamics was applied to probe the gorge fluctuation
behavior of acetylcholinesterase; finite element simulations were used
to connect synaptic geometry with electrophysiological response.
The synapse is the point of communication where a neuron sends its
signal to another cell. The geometries of the synaptic cleft, bounded
by pre- and postsynaptic membranes, differ by muscle type. Synaptic
signal can be carried by the neurotransmitter acetylcholine (ACh), which
is released by the neuron upon arrival of an action potential. ACh diffuses
across the cleft, binds to and activates the postsynaptic receptors,
generating a response in the receiving cell. The enzyme acetylcholinesterase
(AChE) quickly degrades ACh to acetate and choline, deactivating the
receptor and tapering off the response.
The enzyme AChE has several intriguing qualities. First, its key responsibility
of regulating synaptic transmission has made it target of several chemical
agents: from the drugs countering Alzheimer’s disease and myasthenia
gravis (serious muscle weakness), to snake toxins and chemical weapons
(such as sarin and VX). Second, the active site of AChE is buried 2
nm deep in the center of the enzyme, and connected to the protein surface
by a narrow “gorge”. This design is counterintuitive considering
the rapid catalysis of AChE; however, it has been suggested that selective
control of substrate entry may be facilitated by a dynamical, fluctuating
gorge.
We have performed two molecular dynamics simulations, each amounting
to several nanoseconds: one of AChE by itself (10 ns), the other of
AChE with a snake toxin, fasciculin (5 ns). From the static crystallographic
structure, we see that fasciculin obstructs the entrance to the gorge,
but it does not reach down the gorge.
Mechanisms of AChE inhibition by fasciculin. Measuring the width
(“proper radius”) of the gorge every picosecond in the simulations,
we can make a histogram of its distribution (Figure 1). Strikingly,
this is not simply a symmetric, Gaussian distribution; there are two
substates, one wider and one narrower. In addition, fasciculin shifts
the whole distribution towards lower width values, and favors the narrower
substate. These observations lead us to suggest that fasciculin restricts
AChE gorge fluctuation in a dynamical fashion.
Looking at the average structures from the two simulation, we also
noticed that the active site of AChE was changed: the histidine residue
that served as the bridge in the proton-transfer pathway in catalysis
has been oriented away from its normal position. As fasciculin did not
reach down to the active site, this disruption of active site could
only happen by allosteric means.
In sum, fasciculin inhibits AChE by three mechanisms: (i) steric obstruction
of the entrance of the gorge; (ii) dynamic restriction of gorge width;
(iii) allosteric disruption of the active site conformation. The first
is a direct observation from the static crystallographic structure;
the latter two are observations from our simulations.
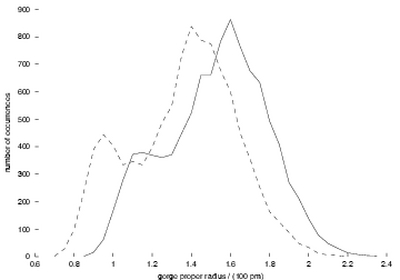
Figure 1:
Distribution of the gorge proper radius. Solid line, from the simulation
of AChE by itself; dashed line, from that of AChE with fasciculin.
“Porcupine plots”: visualizing concerted motions in AChE
gorge fluctuation. What controls the fluctuation of the AChE gorge?
Is it merely the movements of a few residues around the gorge bottleneck,
or are there more global motions at work? With the large amount of data
from the simulations, it is hard to determine one way or the other.
We have created a simple device –the porcupine plot– to help
us understand the correlation of motion in different parts of the protein
with a functionally important motion, the gorge width in this case.
By plotting the correlation vector between the movement of each a-carbon
atom and the gorge width, we see the likelihood and direction of concerted
motions of different parts of the protein whenever the gorge becomes
open. We are looking down the gorge in both panels of Figure 2.
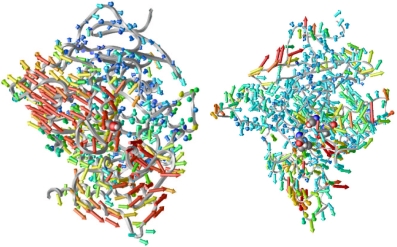
Figure 2:
Porcupine plots. Left, from the simulation of AChE by itself;
right, from that of AChE with fasciculin. The color of each vector
corresponds to the magnitude thereof.
In the left panel, we see that the gorge opening does not only involve
the residues near the gorge, but even residues some distance from the
gorge also move concertedly to make way for the gorge to open. With
fasciculin bound (right panel), much of this coordination has been suppressed;
this may be one of the mechanisms by which fasciculin restricts the
opening of the gorge. The simple device of porcupine plot has proven
to be useful in visualising the concerted motions in proteins.
These nanosecond-scale simulations have enabled us to make comparisons
with experiments. Fluorescence anisotropy decay experiment, a method
detecting fast motions in proteins, has the resolution of nanoseconds.
In cooperation with the Taylor laboratory, we have verified our simulation
results by comparing the decay of anisotropy due to protein segmental
motions calculated from simulation and that observed from experiment.
Going further upward to the organellar scale, we built a robust finite
element software package to solve the time-dependent diffusion equation,
and applied it to the diffusion of the neurotransmitter ACh across the
synaptic cleft. Starting with the geometric shapes of the clefts as
observed from electron microscopy, we calculated the postsynaptic responses
using this package. The simulated response curves were then compared
with electrophysiological data. We reproduced the different response
trends in fast-twitch and slow-twitch muscle synapses.
I have learned this in my doctoral training: We can deepen our understanding
of biomolecular behaviors by applying a combination of physical theory,
computer technology, creative ways of analysis, and careful comparison
with experiments. Computer simulation has opened up exciting ways of
investigating the chemistry in biological systems.
> Kaihsu 's page, including access to full text thesis: <http://sansom.biop.ox.ac.uk/kaihsu/>

