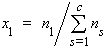
SOLUBILITY OF GASES IN LIQUIDS
The Solubility Data project (SDP) has as its aim a comprehensive review of published data for solubilities of gases, liquids and solids in liquids or solids. Data of suitable precision are compiled for each publication on data sheets in a uniform format. The data for each system are evaluated and, where data from independent sources agree sufficiently, recommended values are proposed. The evaluation sheets, recommended values, and compiled data sheets are published on consecutive pages.
COMPILATIONS AND EVALUATIONS
The formats for the compilations and critical evaluations have been standardized for all volumes. A description of these formats follows.
Compilations
The format used for the compilations is, for the most part, self-explanatory. Normally, a compilation sheet is divided into boxes, with detailed contents described below.
Components: Each component is listed according to IUPAC name, formula, and Chemical Abstracts (CA) Registry Number. The Chemical Abstracts name is also included if this differs from the IUPAC name, as are trivial names if appropriate. IUPAC and common names are cross-referenced to Chemical Abstracts names in the System Index.
The formula is given either in terms of the IUPAC or Hill (1) system and the choice of formula is governed by what is usual for most current users: i.e., IUPAC for inorganic compounds, and Hill system for organic compounds. Components are ordered on a given compilation sheet according to:
In each class, ordering follows the 18-column IUPAC periodic table. The same order is followed in arranging the compilation sheets within a given volume.
Original Measurements: References are abbreviated in the forms given by Chemical Abstracts Service Source Index (CASSI). Names originally in other than Roman alphabets are given as transliterated by Chemical Abstracts. In the case of multiple entries (for example, translations) an asterisk indicates the publication used for compilation of the data.
Variables: Ranges of temperature, pressure, etc. are indicated here.
Prepared by: The names of all compilers are given here.
Experimental Values: Components are described as (1), (2), etc., as defined in the
"Components" box. Data are reported in the units used in the original publication, with the exception that modern names for units and quantities are used; e.g., mass per cent for weight per cent; mol dm-3 for molar; etc. Usually, only one type of value (e.g., mass per cent) is found in the original paper, and the compiler has added the mole fractions from calculations based on 1989 atomic weights (2) and referenced sources of densities, where necessary. Temperatures are expressed as t/°C, t/°F or T/K as in the original; if necessary, conversions to T/K are made, sometimes in the compilations, and always in the critical evaluation. However, the author's units are expressed according to IUPAC recommendations (3) as far as possible.
Errors in calculations, fitting equations, etc. are noted, and where possible corrected. Material inserted by the compiler is identified by the word "compiler" or by the compiler's name in parentheses or in a footnote. Details of smoothing equations (with limits) are included if they are present in the original publication and if the temperature or pressure ranges are wide enough to justify this procedure and if the compiler finds that the equations are consistent with the data.
The precision of the original data is preserved when derived quantities are calculated, if necessary by the inclusion of one additional significant figure. In some cases, compilers note that numerical data have been obtained from published graphs using digitizing techniques. In these cases, the precision of the data can be determined by the quality of the original graph and the limitations of the digitizing technique.
Method: The apparatus and procedure are mentioned briefly. Abbreviations used in Chemical Abstracts are often used here to save space, reference being made to sources of further detail if these are cited in the original paper. Several reviews on experimental methods of determining gas solubilities are given in (4-10).
Source and Purity of Materials: For each component, referred to as (1), (2), etc., the following information (in this order and in abbreviated form) is provided if available in the original paper: source and specified method of preparation; properties; degree of purity. The solubility is usually more sensitive to impurities in the gaseous component than in the liquid component. However, the most important source of impurities is traces of unwanted gas dissolved in the liquid. Inadequate preliminary degassing of the absorbing liquid is probably the most often overlooked serious source of error in gas solubility measurements.
Estimated Error: If estimated errors were omitted by the original authors, and if relevant information is available, the compilers have attempted to estimate errors (identified by "compiler" or the compiler's name in parentheses or in a footnote) from the internal consistency of data and type of apparatus used. Methods used by the compilers for estimating and reporting errors are based on Ku and Eisenhart (11).
Comments and/or Additional Data: Compilations may include this section, in which short comments relevant to the general nature of the work or additional experimental and thermodynamic data are included which are judged by the compiler to be of value to the reader.
References: The format for these follows the format for the Original Measurements box, except that final page numbers are omitted. References (usually cited in the original paper) are given where relevant to interpretation of the compiled data, or where cross-reference can be made to other compilations.
Evaluations
The evaluator's task is to assess the reliability and quality of the data, to estimate errors where necessary, and to recommend "best" values. The evaluation takes the form of a summary in which all the data supplied by the compiler have been critically reviewed. There are only three boxes on a typical evaluation sheet, and these are described below.
Components: The format is the same as on the Compilation sheets.
Evaluator: Name and affiliation of the evaluator(s); date up to which the literature was checked.
Critical Evaluation:
(a) Critical text. The evaluator checks that the compiled data are correct, assesses their reliability and quality, estimates errors where necessary, and recommends numerical values based on all the published data (including theses, reports and patents) for each given system. Thus, the evaluator reviews the merits or shortcomings of the various data. Only published data are considered. Documented rejection of some published data may occur at this stage, and the corresponding compilations may be removed.
The solubilities in comparatively few systems are known with sufficient accuracy to enable a set of recommended values to be presented, either for measurements near atmospheric pressure or at high pressures. Although many systems have been studied by at least two independent groups of workers, the range of pressures or temperatures is often sufficiently different to make meaningful comparison impossible.
Occasionally, it is not clear why two groups of workers obtained very different but internally consistent sets of results at the same temperature and pressure, although both sets were obtained by reliable methods. In such cases, a decisive assessment may not be possible. In some cases, two or more sets of data have been classified as tentative even though the sets are mutually inconsistent.
Many high pressure solubility data have been published in a smoothed form. Such data are particularly difficult to evaluate, and unless specifically discussed by the authors, the estimated error on such values can be regarded only as an "informed guess".
As well, many high pressure solubility data have been obtained in a more general study of high pressure vapor-liquid equilibrium. In such cases a note is included to indicate that additional vapor-liquid equilibrium data are given in the source. Since the evaluation is for the compiled data, it is possible that the solubility data are given a classification which is better than that which would be given for the complete vapor-liquid data (or vice versa). As an example, it is difficult to determine coexisting liquid and vapor compositions near the critical point of a mixture using some common experimental techniques which yield accurate high pressure solubility data. As another example, conventional methods of analysis may give results with an expected error which would be regarded as sufficiently small for vapor-liquid equilibrium data but an order of magnitude too large for acceptable high pressure gas - liquid solubility.
Sometimes it is possible to judge the reliability of data for a particular gas-liquid system by testing whether the data are consistent with the behavior of homologous gases or liquids.
(b) Fitting equations. If the use of a smoothing equation is justifiable the evaluator may provide an equation representing the solubility as a function of the variables reported on all the compilation sheets, stating the limits within which it should be used.
(c) Graphical summary. In addition to (b) above, graphical summaries are often given.
(d) Recommended values. Data are recommended if the results of at least two independent groups are available and they are in good agreement, and if the evaluator has no doubt as to the adequacy and reliability of the applied experimental and computational procedures. Data are reported as tentative if only one set of measurements is available, or if the evaluator considers some aspect of the computational or experimental method as mildly undesirable but estimates that it should cause only minor errors. Data are considered as doubtful if the evaluator considers some aspect of the computational or experimental method as undesirable but still considers the data to have some value where the order of magnitude of the solubility is needed. Data determined by an inadequate method or under ill-defined conditions are rejected. However, references to these data are included in the evaluation together with a comment by the evaluator as to the reason for their rejection.
(e) References. All pertinent references are given here, including all those publications appearing in the accompanying compilation sheets and those which, by virtue of their poor precision, have been rejected and not compiled.
(f) Units. While the original data may be reported in the units used by the investigators, the final recommended values are reported in SI units (3) when the data can be accurately converted.
QUANTITIES AND UNITS USED IN COMPILATION AND EVALUATION OF SOLUBILITY DATA Mixtures, Solutions and Solubilities
A mixture (12) describes a gaseous, liquid or solid phase containing more than one substance, where the substances are all treated in the same way.
A solution (12) describes a liquid or solid phase containing more than one substance, when for convenience one of the substances, which is called the solvent, and may itself be a mixture, is treated differently than the other substances, which are called solutes. If the sum of the mole fractions of the solutes is small compared to unity, the solution is called a dilute solution.
The solubility of a solute 1 (solid, liquid or gas) is the analytical composition of a saturated solution, expressed in terms of the proportion of the designated solute in a designated solvent (13).
"Saturated" implies equilibrium with respect to the processes of dissolution and vaporization; the equilibrium may be stable or metastable. The solubility of a substance in metastable equilibrium is usually greater than that of the same substance in stable equilibrium. (Strictly speaking, it is the activity of the substance in metastable equilibrium that is greater.)
Either point of view, mixture or solution, may be taken in describing solubility. The two points of view find their expression in the reference states used for definition of activities, activity coefficients and osmotic coefficients. Note that the composition of a saturated mixture (or solution) can be described in terms of any suitable set of thermodynamic components.
For gases, the solubility is quoted, where possible, as mole fraction of the saturating gaseous component in the liquid phase at 1 bar partial pressure of gas. The distinction between vapor-liquid equilibria and the solubility of gases in liquids is arbitrary. It is generally accepted that the equilibrium at 300 K between a typical gas such as argon and a liquid such as water is gas liquid solubility whereas the equilibrium between hexane and cyclohexane at 350 K is an example of vapor-liquid equilibrium.
Physicochemical Quantities and Units
Solubilities of gases have been the subject of research for a long time, and have been expressed in a great many ways, as described below. In each case, specification of the temperature and either partial or total pressure of the saturating gaseous component is necessary. The nomenclature and units follow, where possible, ref. (3). A few quantities follow the ISO standards (15) or the German standard (16); see a review by Cvitas (17) for details.
A note on nomenclature. In the IUPAC Green Book (3), the solute is component B and the solvent is component A. In compilations and evaluations, the first-named component (component 1) is the solute, and the second (component 2 for a two-component system) is the solvent. The reader should bear these distinctions in nomenclature in mind when comparing equations given here with those in the Green Book.
1. Mole fraction of substance 1, x1 or x(1) (condensed phases) or y1 (gases):
| eq. [1] | |
where ns is the amount of substance of s, and c is the number of distinct substances present (often the number of thermodynamic components in the system). Mole per cent of substance 1 is 100 x1.
2. Ionic mole fractions of salt i, xi+, xi-:
For a mixture of s binary salts i, each of which ionizes completely into ns+ cations and ns- anions, with ns= ns+ + ns- and a mixture of p non-electrolytes j, of which some may be solvent components, a generalization of the definition in (14) gives:
| eq. [2] | 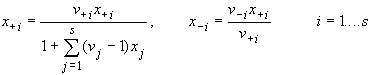 |
| eq. [3] | 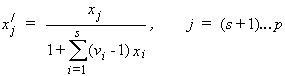 |
The sum of these mole fractions is unity, so that, with c = s + p,
| eq. [4] | 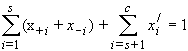 |
General conversions to other units in multicomponent systems are complicated. For a three-component system containing non-electrolyte 1, electrolyte 2 and solvent 3,
| eq. [5] | 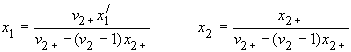 |
These relations are used in solubility equations for salts, and for tabulation of salt effects on solubilities of gases (see below).
3. Mass fraction of substance 1, w1 or w(1):
| eq. [6] | 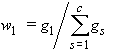 |
where gs is the mass of substance s. Mass per cent of substance 1 is 100 w1. The equivalent terms weight fraction, weight per cent and g (1)/100 g solution are no longer used.
4. Molality of solute 1 in a solvent 2, m1:
| eq. [7] | 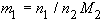 |
SI base units: mol kg-1 . Here, M2 is the molar mass of the solvent. The equivalent term weight solubility, Cw, is no longer used.
5. Amount concentration of solute 1 in a solution of volume V, c1:
| eq. [8] | 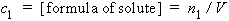 |
SI base units: mol m-3 . The symbol c1 is preferred to [formula of solute], but both are used. The old terms molarity, molar and moles per unit volume are no longer used.
6. Mass concentration of solute 1 in a solution of volume V, ![]() 1 or
1 or ![]() 1:
1:
| eq. [9] | 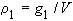 |
SI base units: kg m-3 .
7. Mole ratio, rA,B (dimensionless) (16)
| eq. [10] | 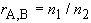 |
Mass ratio, symbol ![]() A,B, may be defined analogously (16).
A,B, may be defined analogously (16).
8. Ionic strength, Im (molality basis), or Ic (concentration basis):
| eq. [11] | 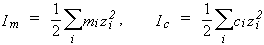 |
where zi is the charge number of ion i. While these quantities are not used generally to express solubilities, they are used to express the compositions of non-saturating components. For a single salt i with ions of charges z+, z-,
| eq. [12] | 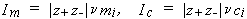 |
Mole and mass fractions are appropriate to either the mixture or the solution point of view. The other quantities are appropriate to the solution point of view only. Conversions between some pairs of these quantities can be carried out using the equations given in Table 1 at the end of this Introduction. Other useful quantities will be defined in the prefaces to individual volumes or on specific data sheets.
In addition to these well-defined SI-based units, other units have been used to express the solubilities of gases. Units and nomenclature follow (4, 5, 7, 18), as modified by IUPAC recommendations (3). The equations describing Bunsen, Kuenen, Ostwald and absorption coefficients, as well as Henry's law constants, hold for ideal gases and perfect solutions only. Corrections for non-ideality should be made where possible. The corrections are less than 1 % for most gases near atmospheric pressure (5).
In much published data, the reference pressure is 1 atm = 0.101325 MPa rather than 1 bar = 0.1 MPa.
9. Bunsen coefficient, ![]() (dimensionless):
(dimensionless):
The volume of saturating gas, V1, reduced to To = 273.15 K, po = 1 bar, which is absorbed by unit volume V2* of pure solvent at the temperature of measurement and partial pressure po = 1 bar. If the gas is ideal, Henry's law (see below) holds, and the liquid is incompressible, then
| eq. [13] | 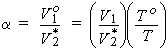 |
10. Kuenen coefficient, S:
The volume of saturating gas, V(g), reduced to To = 273.15 K, po = 1 bar, which is dissolved by unit mass of pure solvent at the temperature of measurement and partial pressure 1 bar. Thus,
| eq. [14] | 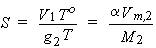 |
SI base units: m3 kg-1 . Here, M2 is the molar mass of the solvent. The Kuenen coefficient is proportional to the the molality of the dissolved gas.
11. Ostwald coefficient, L (dimensionless) (18):
The volume of saturating gas, V1, absorbed by a volume V2* of pure solvent at the temperature and pressure of the measurement. Thus,
| eq. [15] | 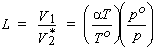 |
The Ostwald coefficient is equal to the ratio of the amount concentrations in the gas and in the liquid.
12. Absorption coefficient, ![]() (dimensionless):
(dimensionless):
The most common of several definitions of absorption coefficient is the volume of gas, reduced to To = 273.15 K, po = 1 bar absorbed per unit volume of pure solvent at a total pressure of 1 bar. The absorption and Bunsen coefficients are therefore very similar, and are connected by
| eq. [16] | 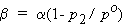 |
where p2 is the partial pressure of the vapor of the solvent.
13. Henry's Law constant, KH:
| eq. [17] | 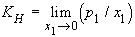 |
SI base units: Pa. Unfortunately, the definition is used often at finite mole fractions, even though this is a limiting law. The following have also been defined as Henry's Law constants:
| eq. [18] | 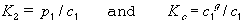 |
where superscript g refers to the gas phase. K2 has SI base units Pa m3 mol-1 , and Kc is dimensionless. The Henry's law constant has also been called the Henry coefficient and the Henry constant. Henry's law can be used, with great caution, to convert data from the experimental pressure to 1 bar if the mole fraction of the gas in the liquid is small, and the difference in pressures is small.
The relations between the mole fraction solubility and the various quantities given above are as follows. Note again that these relations hold for ideal gaseous and perfect solution phases only.
| eq. [19] | 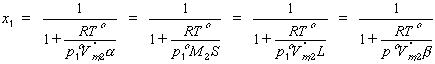 |
14. Salt Effects on the Solubility of Gases (19)
These are often reported as Sechenov (Setchenow, Setschenow) salt effect parameters ksyz, which are defined in various ways. The general semi-empirical Sechenov equation is
| eq. [20] | 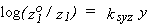 |
where solubility is expressed in quantities z, with superscript o designating pure solvent, and salt composition is expressed in quantities y. The quantities c2, m2, x2', Im and Ic are used for y, and the quantities c1, m1, x1+, a, S and L for z, giving 30 definitions of ksyz. Here, components 1 and 2 are the gaseous solute and electrolyte, respectively. The ratios of z-values are the same for z = c1, a and L and for m1 and S, respectively, leaving 15 distinct definitions. If z is the same, the definitions of ksyz are related simply through 10 equations between pairs of c2, m2, x2', Im and Ic. Some relations among the definitions, in terms of kscc, ksmm and ksxx, are:
| eq. [21] | 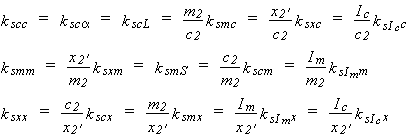 |
These relations hold when a single salt is present; note that the relations between ionic strength and either molality or concentration are simple. If more than one salt is present, the ionic strength is the only practical quantity to be used for y.
Conversions between pairs of kscc, ksmm and ksxx are more complicated, and can be found using eqn [5] and Table 1 at the end of this Introduction. For example,
| eq. [22] | 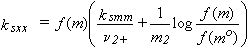 |
where
| eq. [23] | 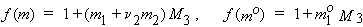 |
Errors in the salt effect parameters, as defined above, can be large. If the relative standard deviation in measurement of solubility is s(c1)/c1, then the relative standard deviation in kscc is
| eq. [24] | 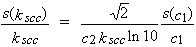 |
For example, for kscc = 0.1 and c2 = 0.01 mol dm-3 , s(kscc)/kscc is 30 % when s(c1)/c1 = 0.05 % and 1200 % when s(c1)/c1 = 2 %. At c2 = 1 mol dm-3 , the corresponding errors are 0.3 and 12 %, respectively.
If the solubility of a gas is greater than about x = 0.01 at partial pressure 1 bar, then several other factors must be taken into account, such as the density of the solution or the partial molar volume of the dissolved gas. In addition, corrections should be made for non-ideality of the gas. See (18) for details.
In addition, the following definitions concerning density are useful in conversions between concentrations and other quantities.
15. Density, ![]() or
or ![]() :
:
| eq. [25] | 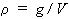 |
SI base units: kg m-3 . Here g is the total mass of the system.
16. Relative density, d = ![]() /
/![]() o : the ratio of the density of a mixture at temperature t, pressure p to the density of a reference substance at temperature t', pressure p'. For liquid solutions, the reference substance is often water at 4°C, 1 bar. (In some cases 1 atm is used instead of 1 bar.) The term specific gravity is no longer used.
o : the ratio of the density of a mixture at temperature t, pressure p to the density of a reference substance at temperature t', pressure p'. For liquid solutions, the reference substance is often water at 4°C, 1 bar. (In some cases 1 atm is used instead of 1 bar.) The term specific gravity is no longer used.
Thermodynamics of Solubility (20)
Thermodynamic analysis of solubility phenomena provides a rational basis for the construction of functions to represent solubility data, and thus aids in evaluation, and sometimes enables thermodynamic quantities to be extracted. Both these aims are often difficult to achieve because of a lack of experimental or theoretical activity coefficients. Where thermodynamic quantities can be found, they are not evaluated critically, since this task would involve examination of a large body of data that is not directly relevant to solubility. Where possible, procedures for evaluation are based on established thermodynamic methods. Specific procedures used in a particular volume will be described in the Preface to that volume.
Only one thermodynamic result is mentioned here: the temperature dependence of solubility. Sometimes it is possible to fit the mole fraction solubility at various temperatures using the equation
| eq. [26] | 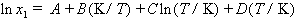 |
where A, B, C and D are constants to be determined from least-squares fitting of the data. Sometimes, to avoid singular matrices of the least-squares normal equations, T is scaled; e.g., T is replaced by T/100.
If the gas and the solution of the dissolved gas are ideal, the coefficients can be used to find standard thermodynamic functions for transfer of the gas from the vapor to the liquid phase at the standard pressure (1 bar) and infinitely-dilute dissolved gas, as follows.
| eq. [27] | 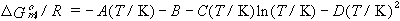 |
| eq. [28] | 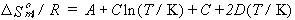 |
| eq. [29] | 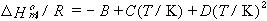 |
| eq. [30] | 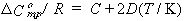 |
Alternatively (but equivalently), the standard state of infinitely-dilute dissolved gas can be described as a hypothetical ideal dissolved gas at mole fraction x1 = 1, p = 1 bar.
REFERENCES
1. Hill, E.A. J. Am. Chem. Soc. 1900, 22, 478.
2. IUPAC Commission on Atomic Weights and Isotopic Abundances. Pure Appl. Chem. 1989, 63, 975.
3. Mills, I.; et al., eds. Quantities, Units and Symbols in Physical Chemistry (the Green Book). Blackwell Scientific Publications. Oxford, UK. 1993.
4. Battino, R.; Clever, H. L. Chem. Rev. 1966, 66, 395.
5. Clever, H. L.; Battino, R. in Solutions and Solubilities (Techniques of Chemistry. Vol. VIII, Part 1). Ed. Dack, M. R. J. J. Wiley & Sons, New York. 1975. Chap. 7.
6. Hildebrand, J. H.; Prausnitz, J. M.; Scott, R. L. Regular and Related Solutions. Van Nostrand Reinhold, New York; 1970; Chap. 8.
7. Markham, A. E.; Kobe, K. A. Chem. Rev. 1941, 28, 519.
8. Wilhelm, E.; Battino, R. Chem. Rev. 1973, 73, 1.
9. Wilhelm, E.; Battino, R.; Wilcock, R. J. Chem. Rev. 1977, 77, 219.
10. Kertes, A. S.; Levy, O.; Markovits, G. Y. in Experimental Thermochemistry, Vol. II, ed. Vodar, B.; LeNaindre, B. Butterworths. London. 1974. Chap. 15.
11. Ku, H.H., p. 73; Eisenhart, C., p. 69; in Ku, H.H., ed. Precision Measurement and Calibration. NBS Special Publication 300. Vol. 1. Washington. 1969.
12. Gold, V.; et al., eds. Compendium of Chemical Technology (the Gold Book). Blackwell Scientific Publications. Oxford, UK. 1987.
13. Freiser, H.; Nancollas, G.H., eds. Compendium of Analytical Nomenclature (the Orange Book). Blackwell Scientific Publications. Oxford, UK. 1987. Sect. 9.1.8.
14. Robinson, R.A.; Stokes, R.H. Electrolyte Solutions. Butterworths. London. 1959. 2nd ed.
15. ISO Standards Handbook, Quantities and Units, International Standards Organization, Geneva, 1993.
16. German Standard, DIN 1310, Zusammensetzung von Mischphasen, Beuth Verlag, Berlin, 1984.
17. Cvitas, T. Chem. International 1995, 17, No. 4, 123.
18. Battino, R. Fluid Phase Equil. 1984, 15, 231.
19. Clever, H.L. in Battino, R. Nitrogen and Air. IUPAC Solubility Data Series, Vol. 10. Pergamon Press. Oxford, UK, 1982. p. xxix.
20. Wilhelm, E. in Battino, R. Nitrogen and Air. IUPAC Solubility Data Series, Vol. 10. Pergamon Press. Oxford, UK, 1982. p. xii.
R. Battino (Dayton, OH, USA)
H.L. Clever (Atlanta, GA, USA)
P.G.T. Fogg (Hertford, UK)
C.L. Young (Melbourne, Australia)
To return to the V.8 home page, use the 'back' feature of your browser or [click here]